Preparing the run
There are 2 files that you have to modify before running your analysis, metadata.tsv and the config yaml file associate withe your sequencing technologies (config_wgbs.yaml or config_nanopore.yaml in the configs folder). The config yaml file is the most important file. It controls the workflow and many tool parameters.
To modify the text files from the terminal you can use vi or nano on iPOP-UP cluster, plus emacs and gedit (the last one being easier to use) on IFB.
Warning
In order to use gedit, be sure that you included -X when connecting to the IFB cluster (-X option is necessary to run graphical applications remotely). See common errors.
Tip
In order to navigate easily in your files with your regular file manager, you can mount your project folder as a disk on your local system. Please follow the instructions for Windows or Linux. This way, you can modify your files directly using any local text editor.
You can also work on your computer and copy the files to the cluster using the scp command or the graphic interface FileZilla.
Danger
Never use word processor (like Microsoft Word or LibreOffice Writer) to modify your code and never copy/past code to/from those softwares. Use only text editors and UTF-8 encoding.
You can find useful help to manage your data on IFB core documentation.
If you're using the IFB Jupyter Hub, it's easier as text and table editors are included, you just have to double click on the file you want to edit, modify and save it using the menu File/Save or Ctrl+S.
metadata.tsv
The experimental description is set up in config/metadata.tsv:
[username@clust-slurm-client Methylator]$ cat configs/metadata.tsv
sample group
SRR9016926 WT
SRR9016927 1KO
SRR9016928 DKO
SRR11806587 WT
SRR11806588 1KO
SRR11806589 DKO
Danger
The columns have to be tab-separated and the header to remain unchanged. Please, don't name groups and samples with an underscore
On Jupyter Hub:
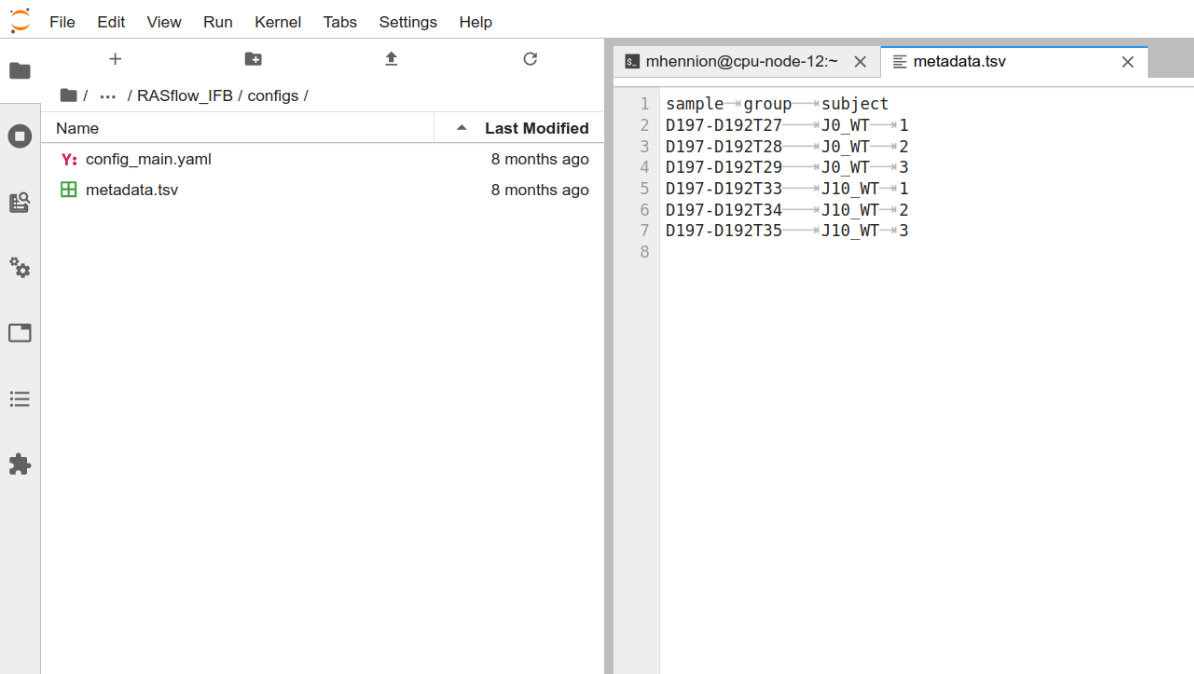
The first column contains the sample names that have to correspond to the FASTQ names that have to correspond to the FASTQ names without the suffix_R1/2.fastq.gz . The second column describes the group the sample belongs to and will be used for differential methylation analysis. You can rename or move that file, as long as you adapt the METAFILE entry in config_main.yaml (see below).
Tip
It is also possible to download and use directly SRA data! That's easy, just enter the SRRxxxx IDs in the first column instead of the sample names! If there is more than one run for your samples, you can also choose to download the SRX directly. The workflow will download all the SSRs associated with each SRX and merge them. Please note that it is not possible to mix SRR and SRX in the metadata !
config_wgbs.yaml
For WGBS or RRBS (Bisulfite conversion + short read sequencing) users must edit the config/config_wgbs.yaml file.
Warning
The yaml format is key:[space]value. The space is mandatory.
This configuration file contains 3 parts:
- Global environment parameters (files and folder path)
- Workflow steps to run and tools parameters
- Specific tools parameters configuration
1) Global environment parameters
Define a project name, paths and shared parameters
[username@clust-slurm-client Methylator]$ cat configs/config_wgbs.yaml
# Project name
PROJECT: Awesome_experience
## paths for intermediate final results
BIGDATAPATH: Big_Data # for big files
RESULTPATH: Results
# ===================== Configuration for BS-seq data =================== #
# ===================================================================== #
DATATYPE: WGBS # WGBS (Whole genome) or "RRBS" (Reduced representation)
METAFILE: /shared/projects/YourProjectName/configs/metadata_wgbs.tsv # the meta file describing the experiment settings
READSPATH: /shared/projects/YourProjectName/fastq # the path to fastq files
COMPARISON: [["WT","1KO"], ["WT","DKO"], ["1KO","DKO"]]
Here you define where the FASTQ files are stored, where is the file describing the experimental set up, the name and localization of the folders where the results will be saved. The results (detailed in results) are separated into two folders:
- the big files: trimmed FASTQ, bam files are in an specific folder defined at BIGDATAPATH
- the small files: QC reports, count tables, BigWig, etc. are in the final result folder defined at RESULTPATH
The small files folder allows for easy retrieval and sharing of results.
Examples are given in the configuration file, but you're free to name and organise them as you want. Be sure to include the full path (starting from /).
Warning
Methylator allows you to performperfom multiple comparisons for experimental design with more than 2 conditions. The reference condition MUST always be in first position eg: KO vs WT differential analysis will be set up as ["WT","KO"]
2) Steps of the workflow you want to run
# ========================= Control of the workflow ========================= #
# =========================================================================== #
## Do you want to download FASTQ files from public from Sequence Read Archive (SRA) ?
SRA: no # "yes" or "no".
## Do you need to do quality control ?
QC: no # "yes" or "no".
## Do you need to do trimming ?
TRIMMED: no # "yes" or "no"
## Do you need to do mapping ?
MAPPING: no # "yes" or "no"
## Do you want to do the globale exploration ?
EXPLORATION: yes # "yes" or "no"
## Do you need to do differentially methylation analysis ?
DIFFERENTIAL: yes # "yes" or "no"
## Do you need a final report ?
REPORT: no # "yes" or "no"
Some default parameters are already chosen. Please read attentively each line and change the value if needed. Please respect the proposed values Dont type YES or Yes or Y but yes for example.
Warning
If QC or SRA is set to yes, the workflow will stop after the QC to let you decide whether you want to trim your raw data or not. In order to run the rest of the workflow, you have to set both QC and SRA to no.
Tip
One might think that a workflow can perform a complete analysis from A to Z in one step (like a pipeline). We don’t think so, at least if you are working with new data. We highly recommend going step by step, carefully checking outputs, QC, and reports in order to select the best parameters for the next steps. The workflow, with its easy setup config file, allows you to explore and dig into your data in an easy and reproducible way. The exploration analysis will provide you with a wide range of stats and graphs to address your experiment. We encourage users to read the original tools documentation to fully understand and control their analysis.
3) Configuration of the specific tools
Here you precise parameters that are specific to one of the steps of the workflow. See detailed description in step by step analysis.
config_nanopore.yaml
The configuration of the workflow for nanopore data.
Warning
The yaml format is key:[space]value. The space is mandatory.
Like the config_wgbs this configuration file contains 3 parts:
1) Define a project name, paths and shared parameters
# Project name
PROJECT: RRMS
## paths for intermediate final results
BIGDATAPATH: Big_Data # for big files
RESULTPATH: Results
## genome files
GENOMEPATH: /shared/projects/edc_nanopore/mm39/mm39.fa # path to the reference genome's folder
## genome
ASSEMBLY: mm39 # mm10 name of the assembly used for the analysis (now use mm39, new version)
# ===================== Configuration for Nanopore data ==================== #
# ========================================================================== #
DATATYPE: NANOPORE # do not touch!
METAFILE: configs/metadata_rrms.tsv
NANO_BAM_PATH: /shared/projects/edc_nanopore/BAM_files/
COMPARISON: [["WT","NP95"]]
# Did you select specific regions during sequencing?
RRMS: yes
BED_RRMS: /shared/projects/edc_nanopore/rrms_mm39_v2_corr_end.bed
5HMC: yes
Warning
if you have perform a RRMS (selection of specific regions during the sequencing), don't forget to put the path to the BED file used for selection below.
Success
Unlike BS-seq sequencing, in nanopore sequencing, it is possible to distinguish between 5mc methylation and 5hmc methylation. If you have basecalled FAST5 files including 5hmC detection, you can explore this mark.
2) Steps of the workflow you want to run
# ========================= Control of the workflow ========================= #
# =========================================================================== #
## Do you want to do the globale exploration ?
EXPLORATION: yes # "yes" or "no"
## Do you need to do differentially methylation analysis ?
DIFFERENTIAL: yes # "yes" or "no"
With nanopore data, the initial steps of the workflow are not necessary. Only the statistical analysis is common to both methods.
3) Configuration of the specific tools
Here you precise parameters that are specific to one of the steps of the workflow. See detailed description in step by step analysis.