Extra help!
How to follow your jobs
Running jobs
You can check the jobs that are running using squeue.
Information about past jobs
The sacct command gives you information about past and running jobs. The documentation is here. You can get different information with the --format option. For instance:
[username@clust-slurm-client Methylator]$ sacct --format=JobID,JobName,Start,CPUTime,MaxRSS,ReqMeM,State
JobID JobName Start CPUTime MaxRSS ReqMem State
------------ ---------- ------------------- ---------- ---------- ---------- ----------
...
9875767 BigWig 2020-07-27T16:02:48 00:00:59 80000Mn COMPLETED
9875767.bat+ batch 2020-07-27T16:02:48 00:00:59 87344K 80000Mn COMPLETED
9875768 BigWigR 2020-07-27T16:02:51 00:00:44 80000Mn COMPLETED
9875768.bat+ batch 2020-07-27T16:02:51 00:00:44 85604K 80000Mn COMPLETED
9875769 PCA 2020-07-27T16:02:52 00:01:22 2000Mn COMPLETED
9875769.bat+ batch 2020-07-27T16:02:52 00:01:22 600332K 2000Mn COMPLETED
9875770 multiQC 2020-07-27T16:02:52 00:01:16 2000Mn COMPLETED
9875770.bat+ batch 2020-07-27T16:02:52 00:01:16 117344K 2000Mn COMPLETED
9875773 snakejob 2020-07-27T16:04:35 00:00:42 2000Mn COMPLETED
9875773.bat+ batch 2020-07-27T16:04:35 00:00:42 59360K 2000Mn COMPLETED
9875774 DEA 2020-07-27T16:05:25 00:05:49 2000Mn RUNNING
Here you have the job ID and name, its starting time, its running time, the maximum RAM used, the memory you requested (it has to be higher than MaxRSS, otherwise the job fails, but not much higher to allow the others to use the resource), and job status (failed, completed, running).
Add -S MMDD to have older jobs (default is today only).
[username@clust-slurm-client Methylator]$ sacct --format=JobID,JobName,Start,CPUTime,MaxRSS,ReqMeM,State -S 0518
Cancelling a job
If you want to cancel a job: scancel job number
Nota: when snakemake is working on a folder, this folder is locked so that you can't start another DAG and create a big mess. If you cancel the main job, snakemake won't be able to unlock the folder (see below).
Having errors?
To quickly check if everything went fine, you have to check the main log. If everything went fine you'll have :
########################################
Date: 2024-05-06T14:51:26+0200
User: username
Host: cpu-node141
Job Name: WGBSflow
Job Id: 32076364
Directory: /shared/projects/wgbs_flow/Elouan/WGBSflow
########################################
WGBS version: v0.1
-------------------------
Main module versions:
Loaded modules : snakemake/7.25.0
conda 23.3.1
Python 3.11.3
apptainer version 1.2.2-1.el7
snakemake
7.25.0
-------------------------
PATH:
/shared/software/conda/envs/snakemake-7.25.0/bin:/shared/software/conda/bin/:/opt/status_bars/status_bars:/opt/conda/bin:/opt/conda/condabin:/opt/status_bars/status_bars:/usr/local/bin:/usr/bin:/usr/local/sbin:/usr/sbin:/shared/home/username/.local/bin:/shared/home/username/bin
########################################
type of data:
wgbs
configs/config_wgbs.yaml
-------------------------
| RUN ID : 20240506T1451 |
-------------------------
Starting WGBS workflow on project: Test_WGBS
Free disk is measured for the cluster project (folder): wgbs_flow
-------------------------
Workflow summary
Is FASTQ quality control required ? no
Is trimming required ? yes
Is mapping required ? yes
Do you want to do the globale exploration ? yes
Do you need to do differentially methylation analysis ? yes
-------------------------
Workflow running....
Starting Trimming...
Nothing to be done (all requested files are present and up to date).
Starting Mapping...
Nothing to be done (all requested files are present and up to date).
Starting exploratory analysis...
Exploratory analysis is done! (0:01:31)
Starting differential analysis...
Differential analysis is done! (0:02:25)
snakemake --profile=configs/ --config time_string=20240506T1451
Starting final report...
The final report is done! (0:00:41)
Exiting...
########################################
---- Errors ----
There were no errors ! It's time to look at your results, enjoy!
########################################
Job finished 2024-05-06T14:56:12+0200
---- Total runtime 286 s ; 4 min ----
If not, you'll see a summary of the errors:
-------------------------
Workflow summary
Is FASTQ quality control required ? no
Is trimming required ? yes
Is mapping required ? yes
Do you want to do the globale exploration ? yes
Do you need to do differentially methylation analysis ? yes
-------------------------
Workflow running....
Starting Trimming...
Trimming is done! (0:00:51)
Starting Mapping...
Error during mapping; exit code: 1
Exiting...
########################################
---- Errors ----
Results/Test_WGBS/logs/20240529T1327_mapping.txt - cd -
Results/Test_WGBS/logs/20240529T1327_mapping.txt - bismark_genome_preparation --bowtie2 --verbose Big_Data/Test_WGBS/mapping_BOWTIE2/bismark_genome
Results/Test_WGBS/logs/20240529T1327_mapping.txt -
Results/Test_WGBS/logs/20240529T1327_mapping.txt -Submitted job 5 with external jobid 'Submitted batch job 37740876'.
Results/Test_WGBS/logs/20240529T1327_mapping.txt -[Wed May 29 13:28:48 2024]
Results/Test_WGBS/logs/20240529T1327_mapping.txt :Error in rule chromsizes:
Results/Test_WGBS/logs/20240529T1327_mapping.txt - jobid: 6
Results/Test_WGBS/logs/20240529T1327_mapping.txt - output: Big_Data/Test_WGBS/mapping_BOWTIE2/chrom.sizes
Results/Test_WGBS/logs/20240529T1327_mapping.txt - shell:
Results/Test_WGBS/logs/20240529T1327_mapping.txt -
Results/Test_WGBS/logs/20240529T1327_mapping.txt - samtools faidx --fai-idx - /shared/projects/wgbs_flow/Elouan/WGBSflow/TestDataset/my_bank/mm39_chr19_mini.fa | cut -f1,2 > Big_Data/Test_WGBS/mapping_BOWTIE2/chrom.sizes
Results/Test_WGBS/logs/20240529T1327_mapping.txt -
Results/Test_WGBS/logs/20240529T1327_mapping.txt - (one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Results/Test_WGBS/logs/20240529T1327_mapping.txt - cluster_jobid: Submitted batch job 37740875
Results/Test_WGBS/logs/20240529T1327_mapping.txt -
Results/Test_WGBS/logs/20240529T1327_mapping.txt :Error executing rule chromsizes on cluster (jobid: 6, external: Submitted batch job 37740875, jobscript: /shared/projects/wgbs_flow/Elouan/WGBSflow/.snakemake/tmp.4q9c7ilv/snakejob.chromsizes.6.sh). For error details see the cluster log and the log files of the involved rule(s).
Results/Test_WGBS/logs/20240529T1327_mapping.txt -[Wed May 29 13:28:49 2024]
Results/Test_WGBS/logs/20240529T1327_mapping.txt :Error in rule genomePrep:
Results/Test_WGBS/logs/20240529T1327_mapping.txt - jobid: 5
Results/Test_WGBS/logs/20240529T1327_mapping.txt - output: Big_Data/Test_WGBS/mapping_BOWTIE2/bismark_genome/Bisulfite_Genome
Results/Test_WGBS/logs/20240529T1327_mapping.txt - shell:
Results/Test_WGBS/logs/20240529T1327_mapping.txt -
Results/Test_WGBS/logs/20240529T1327_mapping.txt - cd Big_Data/Test_WGBS/mapping_BOWTIE2/bismark_genome
--
Results/Test_WGBS/logs/20240529T1327_mapping.txt - bismark_genome_preparation --bowtie2 --verbose Big_Data/Test_WGBS/mapping_BOWTIE2/bismark_genome
Results/Test_WGBS/logs/20240529T1327_mapping.txt -
Results/Test_WGBS/logs/20240529T1327_mapping.txt - (one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Results/Test_WGBS/logs/20240529T1327_mapping.txt - cluster_jobid: Submitted batch job 37740876
Results/Test_WGBS/logs/20240529T1327_mapping.txt -
Results/Test_WGBS/logs/20240529T1327_mapping.txt :Error executing rule genomePrep on cluster (jobid: 5, external: Submitted batch job 37740876, jobscript: /shared/projects/wgbs_flow/Elouan/WGBSflow/.snakemake/tmp.4q9c7ilv/snakejob.genomePrep.5.sh). For error details see the cluster log and the log files of the involved rule(s).
Results/Test_WGBS/logs/20240529T1327_mapping.txt :Exiting because a job execution failed. Look above for error message
Results/Test_WGBS/logs/20240529T1327_mapping.txt -Complete log: .snakemake/log/2024-05-29T132836.854501.snakemake.log
20240529T1327_mapping.txt
Building DAG of jobs...
Using shell: /usr/bin/bash
Provided cluster nodes: 100
Job stats:
job count min threads max threads
----------- ------- ------------- -------------
BamIndex 6 1 1
BedtoBig 6 1 1
GetCoverage 6 1 1
MethExtract 6 1 1
ReadCov 6 1 1
Report 6 1 1
Summary 1 1 1
ToBw 6 1 1
alignmentQC 6 1 1
chromsizes 1 1 1
deduplicate 6 1 1
end 1 1 1
genomePrep 1 1 1
mapping 6 1 1
multiQC 1 1 1
sortBam 6 1 1
total 71 1 1
Select jobs to execute...
[Wed May 29 13:28:38 2024]
rule chromsizes:
output: Big_Data/Test_WGBS/mapping_BOWTIE2/chrom.sizes
jobid: 6
reason: Missing output files: Big_Data/Test_WGBS/mapping_BOWTIE2/chrom.sizes
resources: mem_mb=500, mem_mib=477, disk_mb=1000, disk_mib=954, tmpdir=<TBD>, cpus=1, partition=ipop-up
samtools faidx --fai-idx - /shared/projects/wgbs_flow/Elouan/WGBSflow/TestDataset/my_bank/mm39_chr19_mini.fa | cut -f1,2 > Big_Data/Test_WGBS/mapping_BOWTIE2/chrom.sizes
Submitted job 6 with external jobid 'Submitted batch job 37740875'.
[Wed May 29 13:28:38 2024]
rule genomePrep:
output: Big_Data/Test_WGBS/mapping_BOWTIE2/bismark_genome/Bisulfite_Genome
jobid: 5
reason: Missing output files: Big_Data/Test_WGBS/mapping_BOWTIE2/bismark_genome/Bisulfite_Genome
resources: mem_mb=12000, mem_mib=11445, disk_mb=1000, disk_mib=954, tmpdir=<TBD>, cpus=4, partition=ipop-up
cd Big_Data/Test_WGBS/mapping_BOWTIE2/bismark_genome
ln -s /shared/projects/wgbs_flow/Elouan/WGBSflow/TestDataset/my_bank/mm39_chr19_mini.fa genome.fa
cd -
bismark_genome_preparation --bowtie2 --verbose Big_Data/Test_WGBS/mapping_BOWTIE2/bismark_genome
Submitted job 5 with external jobid 'Submitted batch job 37740876'.
[Wed May 29 13:28:48 2024]
Error in rule chromsizes:
jobid: 6
output: Big_Data/Test_WGBS/mapping_BOWTIE2/chrom.sizes
shell:
samtools faidx --fai-idx - /shared/projects/wgbs_flow/Elouan/WGBSflow/TestDataset/my_bank/mm39_chr19_mini.fa | cut -f1,2 > Big_Data/Test_WGBS/mapping_BOWTIE2/chrom.sizes
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
cluster_jobid: Submitted batch job 37740875
Error executing rule chromsizes on cluster (jobid: 6, external: Submitted batch job 37740875, jobscript: /shared/projects/wgbs_flow/Elouan/WGBSflow/.snakemake/tmp.4q9c7ilv/snakejob.chromsizes.6.sh). For error details see the cluster log and the log files of the involved rule(s).
[Wed May 29 13:28:49 2024]
Error in rule genomePrep:
jobid: 5
output: Big_Data/Test_WGBS/mapping_BOWTIE2/bismark_genome/Bisulfite_Genome
shell:
cd Big_Data/Test_WGBS/mapping_BOWTIE2/bismark_genome
ln -s /shared/projects/wgbs_flow/Elouan/WGBSflow/TestDataset/my_bank/mm39_chr19_mini.fa genome.fa
cd -
bismark_genome_preparation --bowtie2 --verbose Big_Data/Test_WGBS/mapping_BOWTIE2/bismark_genome
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
cluster_jobid: Submitted batch job 37740876
Error executing rule genomePrep on cluster (jobid: 5, external: Submitted batch job 37740876, jobscript: /shared/projects/wgbs_flow/Elouan/WGBSflow/.snakemake/tmp.4q9c7ilv/snakejob.genomePrep.5.sh). For error details see the cluster log and the log files of the involved rule(s).
Exiting because a job execution failed. Look above for error message
Complete log: .snakemake/log/2024-05-29T132836.854501.snakemake.log
You can have the description of the error in the SLURM output corresponding to the external jobid, here 37740875:
[username @ clust-slurm-client Methylator]$ cat slurm_output/chromsizes--37740875.out
Building DAG of jobs...
Using shell: /usr/bin/bash
Provided cores: 2
Rules claiming more threads will be scaled down.
Provided resources: mem_mb=500, mem_mib=477, disk_mb=1000, disk_mib=954, cpus=1
Select jobs to execute...
[Wed May 29 13:28:43 2024]
rule chromsizes:
output: Big_Data/Test_WGBS/mapping_BOWTIE2/chrom.sizes
jobid: 0
reason: Missing output files: Big_Data/Test_WGBS/mapping_BOWTIE2/chrom.sizes
resources: mem_mb=500, mem_mib=477, disk_mb=1000, disk_mib=954, tmpdir=/tmp, cpus=1, partition=ipop-up
samtools faidx --fai-idx - /shared/projects/wgbs_flow/Elouan/WGBSflow/TestDataset/my_bank/mm39_chr19_mini.fa | cut -f1,2 > Big_Data/Test_WGBS/mapping_BOWTIE2/chrom.sizes
Activating singularity image wgbsflow.simg
[E::fai_build3_core] Failed to open the file /shared/projects/wgbs_flow/Elouan/WGBSflow/TestDataset/my_bank/mm39_chr19_mini.fa
[faidx] Could not build fai index -
[Wed May 29 13:28:43 2024]
Error in rule chromsizes:
jobid: 0
output: Big_Data/Test_WGBS/mapping_BOWTIE2/chrom.sizes
shell:
samtools faidx --fai-idx - /shared/projects/wgbs_flow/Elouan/WGBSflow/TestDataset/my_bank/mm39_chr19_mini.fa | cut -f1,2 > Big_Data/Test_WGBS/mapping_BOWTIE2/chrom.sizes
(one of the commands exited with non-zero exit code; note that snakemake uses bash strict mode!)
Removing output files of failed job chromsizes since they might be corrupted:
Big_Data/Test_WGBS/mapping_BOWTIE2/chrom.sizes
Shutting down, this might take some time.
Exiting because a job execution failed. Look above for error message
Common errors
Error starting gedit on IFB
If you encounter an error starting gedit
[unsername @ clust-slurm-client 16:04]$ ~ : gedit toto.txt
(gedit:6625): Gtk-WARNING **: cannot open display:
Be sure to include -X when connecting to the cluster (-X option is necessary to run graphical applications remotely).
Use :
or
You@YourComputer:~$ ssh -X -o "ServerAliveInterval 10" unsername@core.cluster.france-bioinformatique.fr
The option -o "ServerAliveInterval 10" is facultative, it keeps the connection alive even if you're not using your shell for a while.
Initial QC fails
If you don't get MultiQC report_quality_control.html report in results/EXAMPLE/fastqc, you may have some fastq files not fitting the required format:
- SampleName_R1.fastq.gz and SampleName_R2.fastq.gz for pair-end data,
- SampleName.fastq.gz for single-end data.
Please see how to correct a batch of FASTQ files.
Memory
I set up the memory necessary for each rule, but it is possible that big datasets induce a memory excess error. In that case the job stops and you get in the corresponding Slurm output something like this:
slurmstepd: error: Job 8430179 exceeded memory limit (10442128 > 10240000), being killed
slurmstepd: error: Exceeded job memory limit
slurmstepd: error: *** JOB 8430179 ON cpu-node-13 CANCELLED AT 2020-05-20T09:58:05 ***
Will exit after finishing currently running jobs.
In that case, you can increase the memory request by modifying in workflow/resources.yaml the mem entry corresponding to the rule that failed.
[username@clust-slurm-client Methylator]$ cat workflow/resources.yaml
__default__:
mem: 500
name: rnaseq
cpus: 1
qualityControl:
mem: 6000
name: QC
cpus: 2
trim:
mem: 6000
name: trimming
cpus: 8
...
For help you define an adequate reservation of resources (cores and memory).You can use the « Slurm job efficiency report » (seff) that reports on the efficiency of a job’s CPU and memory utilization. Launch the command below, once the execution of the job is complete
(base) [username@ipop-up WGBSflow]$ seff 32076366
Job ID: 32076366
Cluster: production
User/Group: bethuel/umr7216
State: COMPLETED (exit code 0)
Nodes: 1
Cores per node: 4
CPU Utilized: 00:00:55
CPU Efficiency: 24.12% of 00:03:48 core-walltime
Job Wall-clock time: 00:00:57
Memory Utilized: 1.31 GB
Memory Efficiency: 1.12% of 117.19 GB
If the rule that failed is not listed here, you can add it respecting the format. And restart your workflow.
Note
For resource-intensive worklfow steps, if the associated snakemake rules fail, they are automatically restarted, increasing memory by a factor of 2. In all, each rule can be restarted three times consecutively. This prevents the workflow from stopping for resource reasons. Please note, however, that in this case the log files will display error messages every time the rule fails, even if it succeeds with sufficient memory.
Folder locked
When snakemake is working on a folder, this folder is locked so that you can't start another DAG and create a big mess. If you cancel the main job, snakemake won't be able to unlock the folder and next time you run Workflow.sh wgbs, you will get the following error:
Error: Directory cannot be locked. Please make sure that no other Snakemake process is trying to create the same files in the following directory:
/shared/mfs/data/projects/awesome/Methylator
If you are sure that no other instances of snakemake are running on this directory, the remaining lock was likely caused by a kill signal or a power loss. It can be removed with the --unlock argument.
In order to remove the lock, run:
Then you can restart your workflow.
Storage space
Sometimes you may reach the quota you have for your project. To check the quota, run:
In principle it should raise an error, but sometimes it doesn't and it's hard to find out what is the problem. So if a task fails with no error (typically during mapping), try to make more space (or ask for more space on IFB Community support or iPOP-UP Community support) before trying again.
Metadata line break
Lorsque vous compléter le metadat il est possible que des suats de lignes, des erreurs d'indentations ou autre entraine un message d erreur comme celui-çi :
---- Errors ----
Results/MUSCLE_SK_PIG/logs/20240514T1517_fastq_dump_QC.txt -File path Results/MUSCLE_SK_PIG//fastqc/SRA.log contains double '/'. This is likely unintended. It can also lead to inconsistent results of the file-matching approach used by Snakemake.
Results/MUSCLE_SK_PIG/logs/20240514T1517_fastq_dump_QC.txt -File path 'Results/MUSCLE_SK_PIG/fastqc/
Results/MUSCLE_SK_PIG/logs/20240514T1517_fastq_dump_QC.txt -SRR7812212_R1_fastqc.html' contains line break. This is likely unintended. It can also lead to inconsistent results of the file-matching approach used by Snakemake.
Results/MUSCLE_SK_PIG/logs/20240514T1517_fastq_dump_QC.txt -File path 'Results/MUSCLE_SK_PIG/fastqc/
Results/MUSCLE_SK_PIG/logs/20240514T1517_fastq_dump_QC.txt -SRR7812212_R2_fastqc.html' contains line break. This is likely unintended. It can also lead to inconsistent results of the file-matching approach used by Snakemake.
Results/MUSCLE_SK_PIG/logs/20240514T1517_fastq_dump_QC.txt :MissingInputException in rule summaryReport in file /shared/projects/wgbs_flow/Elouan/WGBSflow_1/workflow/fastq_dump_QC.rules, line 111:
Results/MUSCLE_SK_PIG/logs/20240514T1517_fastq_dump_QC.txt -Missing input files for rule summaryReport:
Results/MUSCLE_SK_PIG/logs/20240514T1517_fastq_dump_QC.txt - output: Results/MUSCLE_SK_PIG/fastqc/report_quality_control.html, Results/MUSCLE_SK_PIG/fastqc/report_quality_control_data
Results/MUSCLE_SK_PIG/logs/20240514T1517_fastq_dump_QC.txt - affected files:
Results/MUSCLE_SK_PIG/logs/20240514T1517_fastq_dump_QC.txt - Results/MUSCLE_SK_PIG/fastqc/
Results/MUSCLE_SK_PIG/logs/20240514T1517_fastq_dump_QC.txt -SRR7812212_R1_fastqc.html
Paired-end or single-end ?
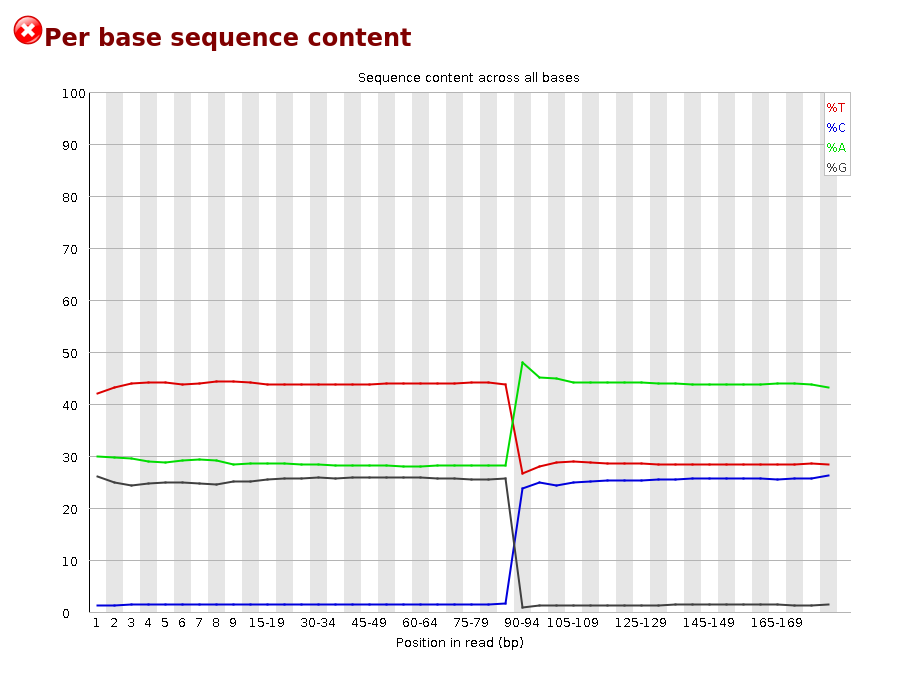
When downloading FASTQ files with the SRA option, errors in the data description are often encountered. The most common one is a confusion between paired-end and single-end data. It frequently happens that sequencing is announced as single-end on SRA but is actually paired-end. In such cases, the "Per Base Sequence Content" graph in the FASTQC file looks like this:
Good practice
- Always save job ID or the dateTtime (ie 20230615T1540) in your notes when launching
Workflow.sh wgbs. It's easier to find the outputs you're interested in days/weeks/months/years later.
Juggling with several projects
If you work on several projects [as defined by cluster documentation], you can either
- have one independant installation of Methylator / project with its own Singularity Image (2 Go). To do that, git clone Methylator repository in each project.
- have an independant Methylator folder, but share the Singularity Image. To do that, git clone Methylator repository in each project, but, before running any analysis, create a symbolic link of wgbsflow.simg from your first project:
[username@clust-slurm-client PROJECT2]$ cd Methylator
[username@clust-slurm-client Methylator]$ ln -s /shared/projects/YourFirstProjectName/Methylator/wgbsflow.simg/ wgbsflow.simg
Tricks
Make aliases
To save time avoiding typing long commands again and again, you can add aliases to your .bashrc file (change only the aliases, unless you know what you're doing).
[username@clust-slurm-client ]$ cat ~/.bashrc
# .bashrc
# Source global definitions
if [ -f /etc/bashrc ]; then
. /etc/bashrc
fi
# Uncomment the following line if you don't like systemctl's auto-paging feature:
# export SYSTEMD_PAGER=
# User specific aliases and functions
alias qq="squeue -u username"
alias sa="sacct --format=JobID,JobName,Start,CPUTime,MaxRSS,ReqMeM,State"
alias ll="ls -lht --color=always"
It will work next time you connect to the server. When you type sa, you will get the command sacct --format=JobID,JobName,Start,CPUTime,MaxRSS,ReqMeM,State running.
Quickly change fastq names
It is possible to quickly rename all your samples using mv. For instance if your samples are named with dots instead of underscores and without R:
[username@clust-slurm-client Raw_fastq]$ ls
D192T27.1.fastq.gz
D192T27.2.fastq.gz
D192T28.1.fastq.gz
D192T28.2.fastq.gz
D192T29.1.fastq.gz
D192T29.2.fastq.gz
D192T30.1.fastq.gz
D192T30.2.fastq.gz
D192T31.1.fastq.gz
D192T31.2.fastq.gz
D192T32.1.fastq.gz
D192T32.2.fastq.gz
You can modify them using mv and a loop on sample numbers.
[username@clust-slurm-client Raw_fastq]$ for i in `seq 27 32`; do mv D192T$i\.1.fastq.gz D192T$i\_R1.fastq.gz; done
[username@clust-slurm-client Raw_fastq]$ for i in `seq 27 32`; do mv D192T$i\.2.fastq.gz D192T$i\_R2.fastq.gz; done
Now sample names are OK: