Workflow architecture
Folders organisation

The configs folder contains all the configurations files.
Files present here are the only ones you need to modify to use the workflow.
.
├── cluster_config_ifb.yaml
├── cluster_config_ipop.yaml
├── cluster_config.yaml
├── config_nanopore.yaml
├── config_wgbs.yaml
├── config.yaml
├── metadata_annot.tsv
└── metadata.tsv
The scripts folder contains all the scripts necessary for the workflow's operation, except for the Snakemake scripts. Please do not touch anything here.
.
├── Annotatr.R
├── build_DAG_graphes.sh
├── check_config_path.py
├── colors.yaml
├── DMR.Rmd
├── DMR_RRBS.Rmd
├── edc_workflows.py
├── final_report_comp.Rmd
├── final_report.Rmd
├── getquota2.sh
├── images
│ ├── bibs_logo_.png
│ ├── cpg_annot.jpeg
│ └── gene.jpeg
├── main_cluster.py
├── MKit_BedgraphDiff.R
├── MKit_Bedgraph.R
├── MKit_BSMAP.R
├── MKit_diff_bed.R
├── Mkit_differential.Rmd
├── MKit_diff_fig.R
├── MKit_Exploration_all.Rmd
├── MKit_Exploration.Rmd
├── MKit_prep_differential.R
├── MKit_prep_nanopore.R
├── MKit_prep_WGBS.R
├── ORA.py
├── parse_yaml.sh
├── parsinglog_flow.py
├── parsinglog.py
├── prep_array.R
├── reporting.py
├── run_rule.sh
├── search_bank.sh
└── test_bam.R
The workflow folder contains all the Snakemake scripts ".rules". Please do not touch anything here.
.
├── config_main_schema.yaml
├── config_mapping_schema.yaml
├── config_methylator_schema.yaml
├── config_QC_schema.yaml
├── config_trim_schema.yaml
├── differential.rules
├── exploration.rules
├── fastq_dump_QC.rules
├── mapping.rules
├── nanopore.yml
├── report.rules
├── samples.schema.yaml
├── Singularity_ncbi
├── trim.rules
└── wgbsflow.yaml
The TestDataset folder contains all the files necessary to test the workflow with a small dataset.
.
├── bam_nanopore
│ ├── RRMS_2marks_NP95
│ └── RRMS_2marks_WT
├── configs
│ ├── config_nanopore.yaml
│ ├── config_wgbs.yaml
│ ├── metadata_nano2.tsv
│ ├── metadata_nano.tsv
│ └── metadata_wgbs.tsv
├── fastq
│ ├── select_sam.sh
│ ├── SRR11806587_sub500000_chr19_R1.fastq.gz
│ ├── SRR11806587_sub500000_chr19_R2.fastq.gz
│ ├── SRR11806588_sub500000_chr19_R1.fastq.gz
│ ├── SRR11806588_sub500000_chr19_R2.fastq.gz
│ ├── SRR11806589_sub500000_chr19_R1.fastq.gz
│ ├── SRR11806589_sub500000_chr19_R2.fastq.gz
│ ├── SRR9016926_sub500000_chr19_R1.fastq.gz
│ ├── SRR9016926_sub500000_chr19_R2.fastq.gz
│ ├── SRR9016927_sub500000_chr19_R1.fastq.gz
│ ├── SRR9016927_sub500000_chr19_R2.fastq.gz
│ ├── SRR9016928_sub500000_chr19_R1.fastq.gz
│ └── SRR9016928_sub500000_chr19_R2.fastq.gz
└── my_bank
├── cpgIslandExt.mm39.bed
├── cpgIslandExt.mm39_mini.bed
├── gencode.vM27.annotation_chr19.gtf
├── gencode.vM27.annotation_chr19_mini.gtf
├── mm39_chr19_mini.fa
└── rrms_mm39_mini.bed
The my_bank folder is an empty directory. It is used to store reference genomes and annotation files (FASTA, GTF, BED, etc.) for different species when the required files are not available in the banks present on your cluster (refer to annotation ).
Folders organisation after run
After launching the workflow, new folders are created. One folder for easily usable results (name by default Results, which you can name as you wish in the configuration file. One folder for 'heavy' results (name by default Big_Data) , also nameable as you wish. A log folder , and a slurm_output folder .
Exemple of folders organisation after launching

The Results folder contains the results of each project. Within each project folder, the results are organized into separate folders corresponding to the different stages of the workflow: fastq download + QC, trimming + QC, mapping + QC, and methylation analysis.
Example of the organization of the results

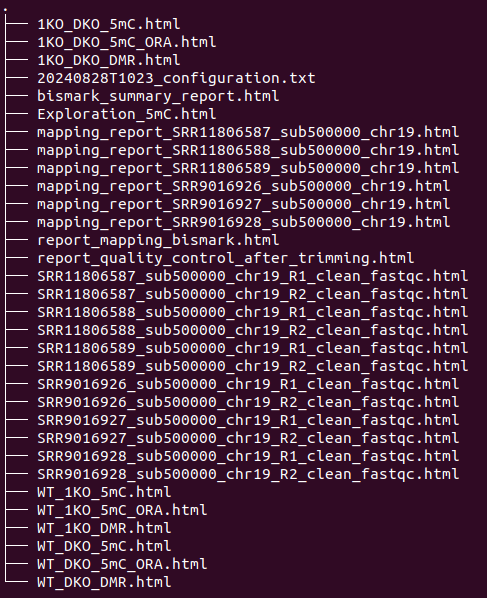
Additionally, at the end of the workflow execution, if you have chosen to generate a report, a zipped and timestamped folder (e.g., final_report_Test_WGBS_20240828T1023.tar.gz) is created to easily share your results. This folder contains all the HTML files (QC + statistical analyses). This folder is accompanied by a timestamped HTML file (e.g., final_report_Test_WGBS_20240828T1023.html), which contains links to the various HTML reports.
Example list of files contained in final_report_(project)_(timestamp).tar.gz
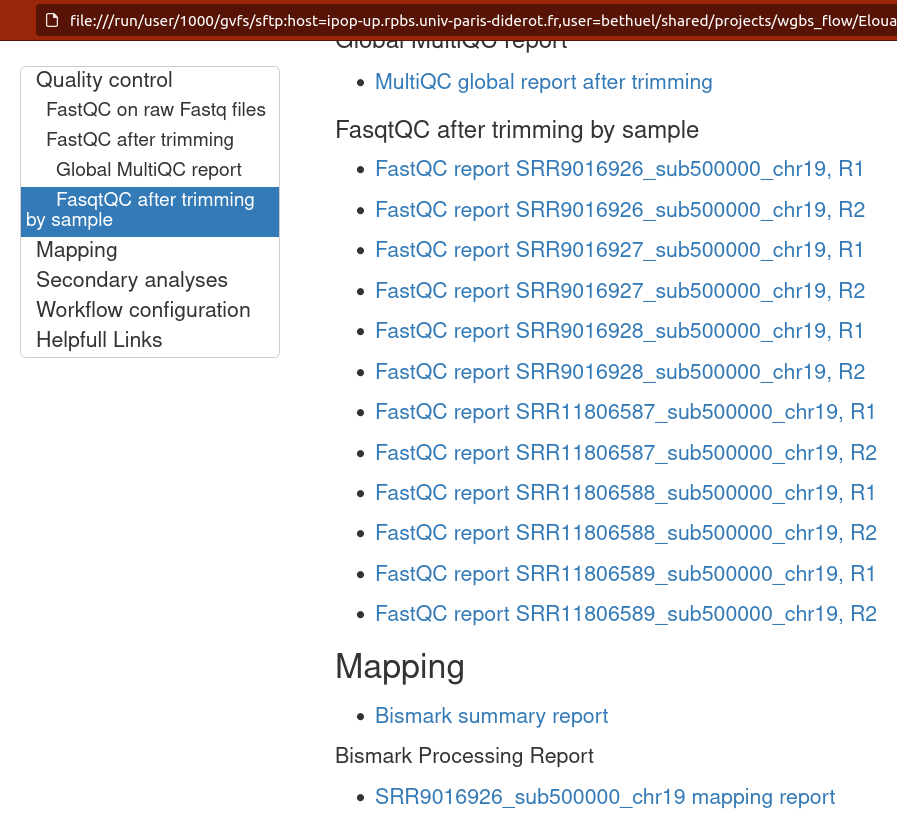
Example of a file: final_report_(project)_(timestamp).html
Tip
The final report folder is unique due to its timestamp, making it easier to version your analyses.
The final report file allows you to navigate more easily through your results.
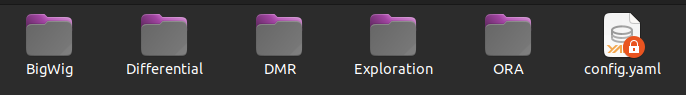
In the Methylation_analysis section of the results, to facilitate the ability to test numerous parameterizations on the same dataset, if you rerun the workflow (for the same project) after modifying a parameter in the Methylation Analysis section of the configuration file, a new methylation analysis is performed. This analysis is stored in the last_analysis folder. For each new analysis, a last_analysis folder is created, and the previous folders are renamed analysis_(number).

To retain information about the parameterization of your analyses, each last_analysis or analysis_(number) folder contains a clone of the configuration file that was used to generate the results. This way, it is possible to conduct multiple methylation analyses without losing track. The methylation analyses are organized according to the different stages of the analyses, namely: exploration Exploration, differential analysis in CpG and Tiles Differential , differential analysis in regions DMR, and enrichment analysis ORA. Additionally, there is a BigWig folder containing bigwig data of the methylation difference (in CpG) for each comparison.